This page describes work done primarily by Gavin Conant, then at the University of New Mexico, with a code from the MIT Whitehead Institute called Genehunter. This work was a collaboration between the UNM Biology Dept, Sandia National Labs, and Pam Fain's group at the University of Colorado. Gavin's participation was funded by DOE thru its computational science graduate fellowship (CSGF) program; Sandia's efforts were funded by DOE's Office of Biological and Environmental Research (OBER).
Genehunter is software package for locating human genetic diseases using linkage analysis. Linkage analysis attempts to locate genes responsible for a disease using genetic data from a family affected by that disease. The genetic profile (or genotype) at a number of markers is determined for as many members of the family as possible. Markers are identifiable locations on a chromosome (often microsatillite positions or single nucleotide polymorphisms) where individual humans are known to show genetic differences. Because the DNA molecule is linear, these markers can be ordered into a list. Each individual has two copies of every marker, one from his or her father and one from his or her mother. Each copy can also be in two possible states, depending on whether the parent transmitted the marker from his or her mother or father (the grandparents of the individual).
To a first approximation, contiguous genes on a chromosome are generally inherited from the same grandparent. However, meiosis, the process of cell division which produces eggs and sperm, can alter this state of affairs. During this process, the parent's two chromosomes can "cross-over", resulting in two new chromosomes which each contain a portion of the original two. Cross-over, also known as recombination, occurs when the phosphate backbones of two DNA helices with similar sequences break and re-join with each other. Because recombination occurs between homologous regions of the original chromosomes, both new chromosomes have a complete set of markers, but the ancestry of these markers is mixed between the grandfather and grandmother of the offspring.
Because such recombination is rare, the closer two markers are on the chromosome the less likely it is that recombination will separate them. Two widely separated markers will almost always have at least one recombination event occur between them and, as a result, will be inherited together only 50% of the time. Thus, the number of recombination events that have occurred between two markers is a distance measure which can be statistically correlated with the pattern of disease incidence in the extended family. This correlation gives insight into where on the chromosome the disease gene is located and is referred to as linkage analysis. Further details of the Genehunter algorithm can be found in the 1987 paper by Lander and Green (below). See the following paper for details on our parallel implementation of Genehunter.
Parallel Genehunter: Implementation of a Linkage Analysis Package for Distributed-Memory Architectures, G. C. Conant, S. J. Plimpton, W. Old, A. Wagner, P. R. Fain, T. R. Pacheco, and G. Heffelfinger, Journal of Parallel and Distributed Computing, 63, 674-682 (2003). (abstract)
The modified GeneHunter code can be downloaded from this page
The original Genehunter project was sponsored by MIT's Whitehead Institute.
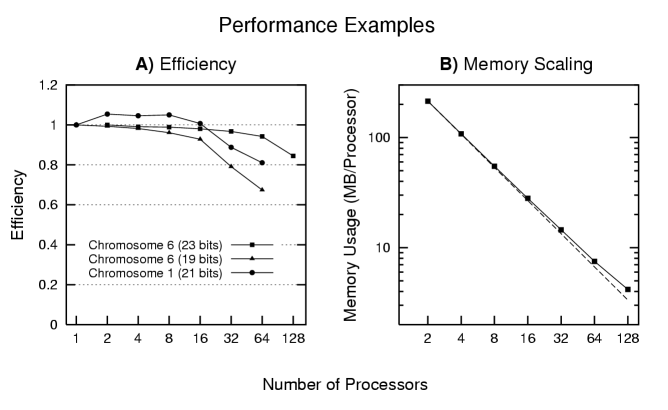
Both memory and computation is distributed in this implementation. The figures below give some indication of the scaling performance of our algorithm for fixed size problems. We computed parallel efficiency for 3 different problem sizes. For reference, the single processor runtimes for the 19 and 21 bit problems are 70 minutes and 38 minutes, respectively. The 23 bit problem will not run on a single processor but requires 34 hours on 2 processors. Panel B gives the memory scaling for this 23 bit problem (the dashed line indicates linear memory scaling).
Genehunter is provided with an example input files (linkloci.dat, linkped.pre). We have provided a batch script for running this input file and a couple of our output for verification purposes.
Gavin Conant is the primary contact for questions and problems with the code. If you cannot reach Gavin, Steve Plimpton can also provide assistance (sjplimp at sandia.gov). We would be delighted to hear if this version of Genehunter is helping with your research and to discuss any ideas you have about the implementation of or modifications to the code.